
Las Anomalías Cromosómicas
Existen diversos tipos de anomalías cromosómicas que requieren el cuidado clínico de un médico o de otro profesional de la salud.
Las anomalías cromosómicas son defectos genéticos que generalmente se producen por desordenes y desbalances en los cromosomas del bebe. Aunque una de las más conocidas sea el síndrome de Down, existen muchas clases de anomalíasAlgunas de estas son menos serias pero otras incluso pueden producir la muerte del niño antes de que nazca. Aproximadamente uno de cada 200 bebés nace con una anomalía cromosómica. Bastantes de los niños con estas anomalías (aunque no todos) se caracterizan por presentar problemas de conducta, retraso mental, incapacidades de aprendizaje, etc. Lo que ocasiona un gran número y tipos de defectos de nacimiento son los errores en la estructura o cantidad de los cromosomas. En ocasiones un infante puede nacer con menos o más cromosomas, o alguno o más rotos o alterados en su estructura.
CAUSAS DE LAS ANOMALÍAS CROMOSÓMICAS
Generalmente, las anomalías cromosómicas se dan por un error durante el desarrollo de una célula espermática u óvulo. Él por qué de estos errores es un misterio. Pero, hasta donde se sabe, nada de lo que haga o deje de hacer cualquiera de los padres antes o durante su desarrollo puede ocasionar una anomalía cromosómica en su hijo. Las células reproductoras(óvulo y célula espermática) tienen solamente 23 cromosomas individuales.
Cuando estas células se unen y empieza el embarazo forman un óvulo fertilizado con 46 cromosomas. Sin embargo aveces algo sale mal antes de que comience el embarazo. En el proceso de división celular, se produce un error que hace que una célula espermática u óvulo termine con un número de cromosomas mayor o menor que lo normal. En el momento en el que esta célula (con una cantidad incorrecta de cromosomas) se une con un óvulo o célula espermática normales, el embrión sufre una anomalía cromosómica. Además a pesar de que una persona tenga la cantidad normal de cromosomas, puede ocurrir que pequeños segmentos de uno o más cromosomas se eliminen, inviertan, dupliquen, se intercambien con parte de otro cromosoma o alteren su ubicación normal.Con frecuencia los embriones que tienen una cantidad incorrecta de cromosomas no sobreviven. En estos casos, la mujer embarazada tiene un aborto espontáneo, casi siempre sin saberlo. Hasta el 70 % de los abortos espontáneos producidos durante el primer trimestre del embarazo se dan por anomalías cromosómicas.
CAUSAS DE LAS ANOMALÍAS CROMOSÓMICAS
Generalmente, las anomalías cromosómicas se dan por un error durante el desarrollo de una célula espermática u óvulo. Él por qué de estos errores es un misterio. Pero, hasta donde se sabe, nada de lo que haga o deje de hacer cualquiera de los padres antes o durante su desarrollo puede ocasionar una anomalía cromosómica en su hijo. Las células reproductoras(óvulo y célula espermática) tienen solamente 23 cromosomas individuales.
Cuando estas células se unen y empieza el embarazo forman un óvulo fertilizado con 46 cromosomas. Sin embargo aveces algo sale mal antes de que comience el embarazo. En el proceso de división celular, se produce un error que hace que una célula espermática u óvulo termine con un número de cromosomas mayor o menor que lo normal. En el momento en el que esta célula (con una cantidad incorrecta de cromosomas) se une con un óvulo o célula espermática normales, el embrión sufre una anomalía cromosómica. Además a pesar de que una persona tenga la cantidad normal de cromosomas, puede ocurrir que pequeños segmentos de uno o más cromosomas se eliminen, inviertan, dupliquen, se intercambien con parte de otro cromosoma o alteren su ubicación normal.Con frecuencia los embriones que tienen una cantidad incorrecta de cromosomas no sobreviven. En estos casos, la mujer embarazada tiene un aborto espontáneo, casi siempre sin saberlo. Hasta el 70 % de los abortos espontáneos producidos durante el primer trimestre del embarazo se dan por anomalías cromosómicas.

¿Qué es un cromosoma?
El cuerpo humano está constituido de células. Por ejemplo, cuando una persona sufre una quemadura solar, la piel se desprende y deja caer "células" de la piel. En el centro de cada célula existe un área llamada núcleo. Los cromosomas humanos se encuentran en el núcleo de la célula. El cromosoma es una estructura que forma parte del núcleo y que contiene los genes. Los genes determinan los rasgos, como el color de ojos y el grupo sanguíneo.
¿Cómo se heredan los cromosomas?
El cuerpo humano está constituido de células. Por ejemplo, cuando una persona sufre una quemadura solar, la piel se desprende y deja caer "células" de la piel. En el centro de cada célula existe un área llamada núcleo. Los cromosomas humanos se encuentran en el núcleo de la célula. El cromosoma es una estructura que forma parte del núcleo y que contiene los genes. Los genes determinan los rasgos, como el color de ojos y el grupo sanguíneo.
¿Cómo se heredan los cromosomas?
Normalmente, cada célula de nuestro cuerpo tiene un total de 46 cromosomas, o 23 pares. Heredamos la mitad de los cromosomas (un miembro de cada par) de nuestra madre biológica y la otra mitad (el miembro homólogo de cada par) de nuestro padre biológico.
Los científicos han enumerado los pares de cromosomas de 1 a 22, habiéndole dado al par 23 el nombre de X o Y, según la estructura. Los primeros 22 pares de cromosomas se llaman "autosomas". Los cromosomas del par 23 se conocen como los "cromosomas sexuales" porque determinan si el bebé será varón o mujer. Las mujeres tienen dos cromosomas "X" y los hombres tienen un cromosoma "X" y un cromosoma "Y". La representación gráfica de los 46 cromosomas, ordenados en pares, recibe el nombre de cariotipo. El cariotipo normal de la mujer se escribe 46, XX, mientras que el cariotipo normal del hombre se escribe 46, XY.
Los científicos han enumerado los pares de cromosomas de 1 a 22, habiéndole dado al par 23 el nombre de X o Y, según la estructura. Los primeros 22 pares de cromosomas se llaman "autosomas". Los cromosomas del par 23 se conocen como los "cromosomas sexuales" porque determinan si el bebé será varón o mujer. Las mujeres tienen dos cromosomas "X" y los hombres tienen un cromosoma "X" y un cromosoma "Y". La representación gráfica de los 46 cromosomas, ordenados en pares, recibe el nombre de cariotipo. El cariotipo normal de la mujer se escribe 46, XX, mientras que el cariotipo normal del hombre se escribe 46, XY.
Los Tipos de Anomalías Cromosómicas
Existen diversos tipos de anomalías cromosómicas que requieren el cuidado clínico de un médico o de otro profesional de la salud.
Existen diversos tipos de anomalías cromosómicas que requieren el cuidado clínico de un médico o de otro profesional de la salud.
Las Anomalías Numéricas: Descripción General de las Trisomías y las Monosomías
¿Qué son las anomalías cromosómicas numéricas?
Las anomalías numéricas conforman uno de los tipos de anomalías cromosómicas. Estos tipos de defectos congénitos ocurren cuando hay un número de cromosomas diferente en las células del cuerpo que el número normal. De modo que, en lugar de los 46 cromosomas habituales en cada célula del cuerpo, hay 45 ó 47 cromosomas. El tener demasiados cromosomas o una cantidad insuficiente de cromosomas constituye una causa para el desarrollo de algún defecto congénito.
¿Qué son las trisomías?
El término "trisomía" se utiliza para describir la presencia de tres cromosomas en lugar del par habitual de cromosomas. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 significa simplemente que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué son las monosomías?
El término "monosomía" se utiliza para describir la ausencia de un miembro de un par de cromosomas. Por lo tanto, habrá un total de 45 cromosomas en cada célula del cuerpo, en lugar de 46. Por ejemplo, si un bebé nace con un solo cromosoma sexual X, en lugar del par habitual (ya sea, dos cromosomas sexuales X o un cromosoma sexual X y un cromosoma sexual Y), se dirá que tiene "monosomía X." La monosomía X también se conoce con el nombre de síndrome de Turner.
¿Qué son las anomalías cromosómicas numéricas?
Las anomalías numéricas conforman uno de los tipos de anomalías cromosómicas. Estos tipos de defectos congénitos ocurren cuando hay un número de cromosomas diferente en las células del cuerpo que el número normal. De modo que, en lugar de los 46 cromosomas habituales en cada célula del cuerpo, hay 45 ó 47 cromosomas. El tener demasiados cromosomas o una cantidad insuficiente de cromosomas constituye una causa para el desarrollo de algún defecto congénito.
¿Qué son las trisomías?
El término "trisomía" se utiliza para describir la presencia de tres cromosomas en lugar del par habitual de cromosomas. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 significa simplemente que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué son las monosomías?
El término "monosomía" se utiliza para describir la ausencia de un miembro de un par de cromosomas. Por lo tanto, habrá un total de 45 cromosomas en cada célula del cuerpo, en lugar de 46. Por ejemplo, si un bebé nace con un solo cromosoma sexual X, en lugar del par habitual (ya sea, dos cromosomas sexuales X o un cromosoma sexual X y un cromosoma sexual Y), se dirá que tiene "monosomía X." La monosomía X también se conoce con el nombre de síndrome de Turner.

Síndrome de Down (Trisomía 21)
¿Qué son las trisomías?
El término "trisomía" se utiliza para describir la presencia de tres cromosomas en lugar del par usual de cromosomas. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 tan solo significa que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué son las trisomías?
El término "trisomía" se utiliza para describir la presencia de tres cromosomas en lugar del par usual de cromosomas. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 tan solo significa que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué es el síndrome de Down?
El síndrome de Down es un trastorno genético que implica una combinación de defectos congénitos, entre los que se incluyen cierto grado de retardo mental, rasgos faciales característicos y, a menudo, defectos cardíacos, deficiencia visual y auditiva y otros problemas de salud. La gravedad de todos estos problemas varía en gran medida entre los individuos afectados. Este síndrome es uno de los defectos genéticos congénitos más comunes y afecta aproximadamente a uno cada 800 a 1000 niños. Según el Instituto Nacional de Síndrome de Down (National Down Syndrome Society), existen más de 350.000 individuos que padecen este síndrome en Estados Unidos. La expectativa de vida de adultos con síndrome de Down es de 55 años aproximadamente, aunque el período de vida promedio varía.
El nombre "síndrome de Down" proviene del médico Dr. Langdon Down, quien fue el primero en describir el conjunto de descubrimientos en 1866. No fue sino hasta 1959 que se identificó la causa del síndrome de Down (la presencia de un cromosoma 21 adicional).
¿Cuáles son las causas del síndrome de Down?
Normalmente en la reproducción, el óvulo de la madre y el espermatozoide empiezan teniendo el número usual de 46 cromosomas. El óvulo y el espermatozoide sufren una división celular en donde los 46 cromosomas se dividen en dos partes iguales y el óvulo y el espermatozoide finalmente poseen 23 cromosomas cada uno. Cuando un espermatozoide con 23 cromosomas fertiliza un óvulo con 23 cromosomas, el bebé tiene finalmente un grupo completo de 46 cromosomas, una mitad obtenida del padre y la otra mitad de la madre.
A veces, ocurre un error mientras los 46 cromosomas se dividen a la mitad y el óvulo o el espermatozoide, en lugar de reservar tan solo una copia del cromosoma 21, sigue teniendo ambas. Si este óvulo o espermatozoide se fertiliza, el bebé acabará teniendo tres copias del cromosoma 21 y esto es lo que se llama "trisomía 21" o síndrome de Down. Las características del síndrome de Down se originan porque cada célula del cuerpo posee una copia adicional del cromosoma 21.
El noventa y cinco por ciento de los casos de síndrome de Down se produce por la Trisomía 21. En algunas ocasiones, el cromosoma 21 adicional, o una porción de ella, se adhiere a otro cromosoma del óvulo o el espermatozoide; esto puede conducir a lo que se denomina síndrome de Down por "translocación" (el 3 a 4 por ciento de los casos). éste es el único tipo de síndrome de Down que puede, a veces, heredarse de alguno de los padres. Algunos padres tienen un reordenamiento que no afecta su salud denominado translocación balanceada, donde el cromosoma 21 se adhiere a otro cromosoma. Con poca frecuencia, tiene lugar un tipo de síndrome de Down llamado el síndrome de Down con alteración cromosómica en "mosaico", cuando ocurre un error en la división celular después de la fertilización (1 a 2 por ciento de los casos). Estas personas tienen algunas células con un cromosoma 21 adicional y otras con el número normal.
¿¿Cuál es el aspecto físico de un niño con síndrome de Down?

Un niño con síndrome de Down puede tener ojos inclinados hacia arriba y orejas pequeñas con la parte superior apenas doblada. La boca puede ser pequeña, lo que hará que la lengua se vea grande. También la nariz puede ser pequeña, con el tabique nasal aplanado. Algunos bebés con síndrome de Down tienen el cuello corto y las manos pequeñas con dedos cortos. En lugar de presentar tres "líneas" en la palma de la mano, el niño con síndrome de Down generalmente tiene una sola línea que atraviesa toda la palma y una segunda línea que forma una curva al lado del pulgar. Con frecuencia, el niño o adulto con síndrome de Down es de baja estatura y las articulaciones son inusualmente laxas. La mayoría de los niños con síndrome de Down presenta algunos de estas características, si bien no todas.
¿Qué clase de problemas tienen generalmente los niños con síndrome de Down?
Alrededor del 40 a 50 por ciento de los niños con síndrome de Down tienen defectos cardíacos. Algunos defectos son menores y se los puede tratar con medicamentos, mientras que otros necesitan una cirugía. Todos los niños con síndrome de Down deben ser examinados por un cardiólogo pediátrico, un médico que se especializa en cardiopatías de niños. Para que cualquier clase de cardiopatía pueda ser tratada, durante los dos primeros meses de vida el médico deberá realizar al niño un ecocardiograma (un procedimiento que evalúa la estructura y el funcionamiento del corazón utilizando ondas sonoras que se registran en un sensor electrónico, produciendo así una imagen en movimiento del corazón y de las válvulas del corazón).
Alrededor del 10 por ciento de los niños con síndrome de Down nace con malformaciones intestinales que necesitan cirugía.
Los niños con síndrome de Down tienen un mayor riesgo de deficiencia visual o auditiva. Entre los problemas visuales más comunes se incluyen la bizquera, la miopía o hipermetropía y las cataratas. La mayoría de los problemas en la visión pueden mejorarse con anteojos, cirugía u otros tratamientos. Durante el primer año de vida, se debe consultar a un oftalmólogo pediátrico (un médico especializado en el cuidado completo de los ojos que examina, diagnostica y define el tratamiento para una variedad de trastornos oculares).
Los niños con síndrome de Down pueden presentar una pérdida de la audición debida a la presencia de líquido en el oído medio, un defecto nervioso o ambos. Todos los niños con síndrome de Down deben someterse a exámenes de visión y audición regularmente de manera que cualquier problema pueda ser tratado antes de que dificulte el desarrollo del lenguaje y otras habilidades.
Los niños con síndrome de Down tienen un mayor riesgo de padecer problemas de tiroides y leucemia. También suelen contraer resfríos, bronquitis y neumonía con frecuencia. Los niños con síndrome de Down deben recibir atención médica con regularidad incluyendo vacunaciones infantiles. El Congreso Nacional de Síndrome de Down (National Down Syndrome Congress) publica una "Lista de Verificación de Medicina Preventiva" (Preventative Medicine Checklist) en la que se encuentran los exámenes médicos recomendados para las diferentes edades.
¿Cuán serio es el retardo mental que se presenta con el síndrome de Down
El grado de retardo mental que se presenta con el síndrome de Down varía enormemente, oscilando desde leve a moderado a severo. Sin embargo, el retardo mental más común es de leve a moderado. No existe la manera de predecir el desarrollo mental del niño con síndrome de Down basándose en los rasgos físicos.
¿Qué puede hacer un niño con síndrome de Down?
Los niños con síndrome de Down generalmente pueden realizar la mayoría de las actividades que cualquier niño realiza, como caminar, hablar, vestirse y aprender a usar el baño. Sin embargo, ellos generalmente hacen estas cosas más tarde que
 los otros niños. No se puede predecir la edad precisa en que se alcanzarán estos logros en el desarrollo. No obstante, los programas de intervención temprana, a partir de la primera infancia, pueden ayudar a estos niños a alcanzar su potencial individual.
los otros niños. No se puede predecir la edad precisa en que se alcanzarán estos logros en el desarrollo. No obstante, los programas de intervención temprana, a partir de la primera infancia, pueden ayudar a estos niños a alcanzar su potencial individual.¿Puede asistir a la escuela un niño con síndrome de Down?
Sí. Existen programas especiales que comienzan en los años de la educación preescolar para ayudar a que los niños con síndrome de Down desarrollen sus habilidades tanto como les sea posible. Además de beneficiarse con una intervención temprana y una educación especial, muchos niños pueden, hasta cierto punto, ser integrados en el aula de clases común. La perspectiva para estos niños es mucho más alentadora de lo que alguna vez lo fue. Muchos aprenderán a leer y escribir y participarán en diferentes actividades para niños ya sea en la escuela o en los vecindarios. Si bien existen programas especiales de trabajo diseñados para adultos con síndrome de Down, muchas personas con el trastorno pueden tener trabajos comunes. En la actualidad, un número cada vez mayor de adultos con síndrome de Down vive de manera semi-independiente en casas de grupos de comunidad en donde cuidan de sí mismos, participan de las tareas domésticas, establecen vínculos de amistad, comparten actividades de tiempo libre y trabajan en su comunidad.
Sí. Existen programas especiales que comienzan en los años de la educación preescolar para ayudar a que los niños con síndrome de Down desarrollen sus habilidades tanto como les sea posible. Además de beneficiarse con una intervención temprana y una educación especial, muchos niños pueden, hasta cierto punto, ser integrados en el aula de clases común. La perspectiva para estos niños es mucho más alentadora de lo que alguna vez lo fue. Muchos aprenderán a leer y escribir y participarán en diferentes actividades para niños ya sea en la escuela o en los vecindarios. Si bien existen programas especiales de trabajo diseñados para adultos con síndrome de Down, muchas personas con el trastorno pueden tener trabajos comunes. En la actualidad, un número cada vez mayor de adultos con síndrome de Down vive de manera semi-independiente en casas de grupos de comunidad en donde cuidan de sí mismos, participan de las tareas domésticas, establecen vínculos de amistad, comparten actividades de tiempo libre y trabajan en su comunidad.
¿Se pueden casar las personas con síndrome de Down?
Algunas personas con síndrome de Down se casan. Aunque ha habido raras excepciones, los hombres con síndrome de Down no pueden engendrar hijos. Durante una gestación, la mujer con síndrome de Down tiene una posibilidad de 50/50 de concebir un niño con síndrome de Down, pero muchas gestaciones se pierden en abortos espontáneos.
¿Cómo se diagnostica el síndrome de Down?
Ya que el índrome de Down está formado por un conjunto tan particular de características, los médicos a veces pueden determinar la existencia del síndrome en un niño solamente con un examen físico. Para confirmar los hallazgos del examen físico, se puede tomar una pequeña muestra de sangre y analizar los cromosomas para determinar la presencia de material de cromosoma 21 adicional. Esta información es importante a la hora de determinar el riesgo en futuras gestaciones. (El síndrome de Down por translocación y el síndrome de Down con alteración cromosómica en mosaico tienen riesgos de recurrencia diferentes).
Las anomalías cromosómicas, como el síndrome de Down, pueden diagnosticarse frecuentemente antes del nacimiento a través del análisis de las células del líquido amniótico o de la placenta. La ecografía fetal durante el embarazo también puede proporcionar información sobre la posibilidad del síndrome de Down, pero la ecografía no tiene una precisión del 100 por ciento, dado que muchos bebés con síndrome de Down presentan en la ecografía el mismo aspecto que un bebé sin síndrome de Down. Un análisis cromosómico, ya sea de una muestra de sangre o de células provenientes del líquido amniótico o de la placenta, tiene una exactitud superior al 99,9 por ciento.
¿Cuáles son los riesgos respecto a la edad materna para el síndrome de Down?
La Edad Materna
El Riesgo en el Nacimiento
25 a 29 años
1 de cada 1250
35 años
1 de cada 378
40 años
1 de cada 100
45 años (o mayor)
1 de cada 30
¿Cuál es el riesgo de que los padres de un niño con síndrome de Down tengan otro niño con síndrome de Down?
En general, para las mujeres menores de 40, luego de haber tenido un bebé con síndrome de Down, la probabilidad de tener otro bebé con síndrome de Down es del 1 por ciento. También se sabe que la posibilidad de síndrome de Down se incrementa con la edad de la madre y, después de los 40 años, una madre estaría en riesgo simplemente basándose en su edad en el momento del parto. Es importante saber que cerca del 80 por ciento de los bebés con síndrome de Down nacen de mujeres menores de 35 años. Esto se debe a que las mujeres menores de 35 años tienen más bebés que las mujeres mayores de 35. El médico puede derivar a los padres a un genetista o a un asesor genético, quien puede explicar los resultados de los exámenes cromosómicos en detalle, así como los riesgos de reincidencia en otro embarazo y los exámenes disponibles para el diagnóstico de problemas cromosómicos antes del nacimiento del bebé.
¿Se puede curar o prevenir el síndrome de Down?
No existe cura para el síndrome de Down. No hay certezas acerca de cómo prevenir el error cromosómico que provoca el síndrome de Down. Hasta la fecha, no existen razones para creer que el padre podría haber hecho algo para provocar o prevenir el nacimiento de su hijo con síndrome de Down. Sin embargo, una investigación reciente sugiere que algunas mujeres que han tenido un hijo con síndrome de Down presentaban una anomalía en la manera en que sus cuerpos metabolizaban (procesaban) el ácido fólico vitamina B. De confirmarse, este hallazgo puede ser incluso otra razón por la que todas las mujeres que estén por concebir deben tomar una multivitamina diaria con 400 microgramos de ácido fólico (del cual se ha demostrado que reduce el riesgo de algunos defectos congénitos del cerebro y la médula espinal).
Algunos afirman que suministrar diferentes vitaminas en altas dosis a los niños con síndrome de Down mejora el rendimiento mental y reduce el retardo mental. Sin embargo, hasta la fecha, no se han realizado investigaciones médicas para comprobar que esto sea realmente así. Es importante que las nuevas familias hablen con el médico, con otras familias y con el Congreso Nacional de Síndrome de Down (National Down Syndrome Congress) con el fin de conocer a qué debe estar preparada ante la presencia del síndrome de Down y conocer las cosas que puedan resultarle útiles al criar un niño con síndrome de Down.
¿Qué investigaciones se están conduciendo acerca del síndrome de Down?
La entidad March of Dimes está investigando las causas de los errores en la división cromosómica, con la esperanza de algún día llegar a prevenir el síndrome de Down y otros defectos congénitos provocados por anomalías en el número o la estructura de los cromosomas. Otros investigadores están buscando mejorar la perspectiva para los niños con síndrome de Down. Un ejemplo es el desarrollo de mejores programas de intervención en el lenguaje para contribuir a que estos niños se comuniquen más fácilmente.

Trisomía 18 y 13
¿Qué son las trisomías?
El término trisomía se utiliza para describir la presencia de tres cromosomas, en lugar del par de cromosomas habitual. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 significa simplemente que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué son las trisomías?
El término trisomía se utiliza para describir la presencia de tres cromosomas, en lugar del par de cromosomas habitual. Por ejemplo, si un niño nace con tres cromosomas 21 en lugar del par usual, se diría que posee "trisomía 21". La trisomía 21 también se conoce como síndrome de Down. Otros ejemplos de trisomía incluyen la trisomía 18 y la trisomía 13. Nuevamente, trisomía 18 o trisomía 13 significa simplemente que existen tres copias y no el par usual del cromosoma 18 (o del cromosoma 13) en cada célula del cuerpo.
¿Qué son la trisomía 18 y la trisomía 13?
La trisomía 18 y la trisomía 13 son trastornos genéticos que presentan una combinación de defectos congénitos que incluyen retardo mental grave, así como problemas de salud que comprometen a casi todos los sistemas orgánicos del cuerpo. Desafortunadamente, 90 por ciento de los recién nacidos con trisomía 18 o 13 mueren por edad 1. Es importante destacar que entre el 5 y el 10 por ciento de los bebés con trisomía sobreviven al primer año de vida. Por lo tanto, estos trastornos no son fatales en todos los casos y, ante la ausencia de problemas inmediatos que pongan en peligro la vida, es difícil hacer predicciones precisas respecto de la expectativa de vida. Existen algunos informes sobre bebés con trisomía 18 o 13 que sobrevivieron hasta la adolescencia. Sin embargo, estos casos son poco frecuentes.
La trisomía 18 se denomina también "síndrome de Edwards", llamada así por el primer médico que describió el trastorno. La trisomía 13 se denomina "síndrome de Patau", en honor de el primer médico que describió el trastorno.
¿Cuáles son las causas de la trisomía 18 y la trisomía 13?
En general cada óvulo y cada espermatozoide contiene 23 cromosomas. La unión de estos crea 23 pares, o 46 cromosomas en total, cuando se realiza la fecundación. De esta manera, una persona recibe exactamente la mitad de su material genético de cada uno de los padres. En ocasiones, ocurre un error durante la formación del óvulo o del espermatozoide, y esto causa la presencia de un cromosoma 18 o 13 adicional. Cuando esta célula aporta el cromosoma 18 adicional al embrión, el resultado es la trisomía 18. Cuando esta célula otorga el cromosoma 13 adicional al embrión, el resultado es la trisomía 13. El cromosoma 18 o 13 adicional puede provenir tanto del óvulo de la madre como del espermatozoide del padre. Las características de la trisomía 18 y la trisomía 13 son el resultado de la presencia de este cromosoma 18 o 13 adicional en cada célula del cuerpo.
En algunas ocasiones, el cromosoma 18 o 13 adicional se adhiere a otro cromosoma en el óvulo o el espermatozoide; esto se denomina translocación. ésta es la única forma de trisomía 18 o 13 que puede ser hereditaria. A veces, uno de los padres puede portar un reordenamiento "balanceado" en el cual el cromosoma 18 o 13 se adhiere a otro cromosoma. Sin embargo, como el padre no tiene ningún material cromosómico adicional o ausente, se dice que presenta una "translocación balanceada" y suele ser normal y goza de buena salud. Esporádicamente, puede ocurrir una trisomía 18 o 13 en mosaico cuando el error en la división celular ocurre después de la fecundación. Estas personas poseen algunas células con un cromosoma 18 o 13 adicional y otras con la cantidad normal.
¿Qué clase de problemas tienen generalmente los niños con trisomía 18 o trisomía 13?
Bebés con trisomía 18Los bebés con trisomía 18 son delgados y frágiles. No pueden desarrollarse y padecen problemas de alimentación. La trisomía 18 provoca microcefalia, con la parte posterior de la cabeza (occipucio) prominente. Las orejas suelen encontrarse más abajo de lo normal en la cabeza. La boca y la mandíbula suelen ser pequeñas y el esternón (hueso del tórax) es más corto.Cuando nacen, estos bebés son pequeños para su edad, aun cuando hayan nacido en término, y poseen un llanto débil. Presentan una disminución en la respuesta al sonido y suelen existir antecedentes de actividad fetal poco frecuente durante la gestación. Alrededor del 90 por ciento de los bebés con trisomía 18 poseen defectos cardíacos. Aprietan los puños de una manera característica y les resulta dificultoso extender los dedos por completo. Suelen presentar contracturas en las articulaciones - donde los brazos y las piernas se encuentran flexionados en lugar de extendidos. Los pies pueden llamarse "de base redondeada" debido a su forma.Los bebés con trisomía 18 también pueden presentar espina bífida, problemas oculares, labio leporino y paladar hendido y pérdida de la audición (en la mayoría de los casos). También son frecuentes los problemas para alimentarse, el crecimiento lento, las convulsiones, presión sanguínea alta, problemas renales y escoliosis (curvatura de la columna vertebral). En los varones, los testículos no descienden al escroto.La mayoría de los bebés con trisomía 18 presenta problemas que afectan a todas las partes del cuerpo en algún grado. La mayoría de los niños con trisomía 18 presentará alguno de los problemas de salud, pero no todos, mencionados anteriormente. Los problemas cardíacos, las dificultades en la alimentación y la mayor susceptibilidad a contraer infecciones son factores que, con frecuencia, contribuyen a la muerte de estos niños.
Bebés con trisomía 13Los bebés con trisomía 13 suelen tener un bajo peso al nacer, aun cuando hayan nacido en término. Presentan microcefalia, con una frente prominente. En general, existen problemas estructurales importantes en el cerebro que se diagnostican al poco tiempo del nacimiento. Con frecuencia, la parte frontal del cerebro no se divide correctamente, lo que resulta en un trastorno denominado holoprosencefalia. Esto puede producir cambios en el desarrollo del rostro del bebé, donde los ojos se encuentran muy juntos, o la nariz o las fosas nasales no se desarrollan completamente. El labio leporino y el paladar hendido se presentan en un 60 a un 80 por ciento de los bebés con trisomía 13.Son frecuentes los problemas oculares y las orejas se encuentran ubicadas más abajo de lo normal y poseen una forma poco común. A veces, los bebés con trisomía 13 pueden presentar anomalías en el cuero cabelludo (aplasia cutis) que se asemejan a las úlceras. También pueden presentar marcas de nacimiento que son de un color rojo purpúreo, formadas por vasos sanguíneos diminutos ubicados cerca de la piel (hemangiomas).Muchos bebés con trisomía 13 presentan dedos adicionales en las manos y en los pies (polidactilia). Los pies pueden presentar talones prominentes. En muchos casos, existen otros problemas de salud presentes al nacer. Estos incluyen defectos cardíacos, problemas renales y,o un onfalocele (un trastorno en el cual algunos de los órganos abdominales sobresalen a través de un orificio en los músculos abdominales en la zona del cordón umbilical). En los varones, a veces los testículos no descienden al escroto. Las mujeres pueden presentar un útero deforme, llamado útero bicorne.
¿Cómo se diagnostican las trisomías 18 y 13?
Como la trisomía 18 y la trisomía 13 poseen cada una su grupo de características específicas, un médico puede determinar si un bebé presenta trisomía 18 o 13 con sólo realizar un examen físico. Para confirmar los resultados del examen físico, puede tomarse una pequeña muestra de sangre y analizar los cromosomas para determinar la presencia del cromosoma 18 o 13 adicional.
Las anomalías cromosómicas, como la trisomía 18 y 13, pueden diagnosticarse antes del nacimiento a través del análisis de las células del líquido amniótico o de la placenta. La ecografía fetal durante el embarazo también puede proporcionar información sobre la posibilidad de la trisomía 18 o 13, pero la ecografía no tiene una precisión del 100 por ciento, dado que algunos bebés con trisomía 18 y 13 presentan en la ecografía el mismo aspecto que un bebé sin el síndrome. Un análisis cromosómico, ya sea de una muestra de sangre o de células provenientes del líquido amniótico o de la placenta, tiene una exactitud superior al 99,9 por ciento.
¿Cuál es el riesgo de que los padres de un niño con trisomía 18 o trisomía 13 tengan otro bebé con trisomía 18 o 13?
En general, en cada embarazo posterior, la probabilidad de tener otro bebé con trisomía 18 o 13 no supera el 1 por ciento. El riesgo de tener un bebé con trisomía 18 o 13 aumenta levemente con relación a la edad de la madre.
Después del nacimiento, el médico suele extraer una muestra de sangre del bebé que podría padecer trisomía 18 o 13 para realizar un análisis cromosómico (denominado cariotipo). éste confirma los hallazgos del médico de la presencia de trisomía 18 o 13 y determina la anomalía cromosómica subyacente. Esta información es importante a la hora de determinar el riesgo en futuras gestaciones. (La translocación y la trisomía 18 y 13 en mosaico poseen diferentes grados de riesgo de reincidencia). El médico puede derivar a los padres a un genetista o a un asesor genético, quien puede explicar los resultados de los exámenes cromosómicos en detalle, así como los riesgos de reincidencia en otro embarazo y los exámenes disponibles para el diagnóstico de problemas cromosómicos antes del nacimiento del bebé.

¿Se puede curar o prevenir la trisomía 18 o 13?
No existe cura para la trisomía 18 o 13. Se desconoce cómo prevenir el error cromosómico que causa la trisomía 18 y la trisomía 13. Hasta este momento, no existe evidencia científica que demuestre que un padre puede haber hecho algo para causar o prevenir el nacimiento de su bebé con trisomía 18 o 13.
¿Qué más debería saber acerca de la trisomía 18 y la trisomía 13?
Debido a que muchos bebés que nacen con trisomía 18 y trisomía 13 pueden vivir sólo unos pocos días o semanas, es posible que los padres deban enfrentar el hecho de que su bebé nunca salga del hospital. Los médicos y los padres necesitan discutir abiertamente acerca de los procedimientos de cuidados intensivos correctos e incorrectos.
Muchas veces los padres están asustados y agobiados por toda la información asociada con las trisomías 18 y 13. Las decisiones que rodean al cuidado del bebé con trisomías 18 y 13 son difíciles y personales. Existen muchos recursos disponibles para ayudar a los padres durante este período, como los servicios de intervención precoz, los cuidados de hospicio, los trabajadores sociales, el capellán o sacerdote del hospital y los asesores genéticos. Las familias que tienen o tuvieron un bebé con trisomía 18 o trisomía 13 suelen ser de gran ayuda y apoyo ya que han experimentado muchas de las mismas dudas y emociones.
¿Dónde pueden obtener información las familias afectadas por la trisomía 18 y la trisomía 13?
Existen organizaciones en todo el país que brindan información y apoyo a las familias con niños afectados por la trisomía 18 y la trisomía 13.
Síndrome de Turner
¿Qué son las monosomías?
El término monosomía se utiliza para describir la ausencia de uno de los miembros de un par de cromosomas. Por lo tanto, habrá un total de 45 cromosomas en cada célula del cuerpo, en lugar de 46. Por ejemplo, si un bebé nace con un solo cromosoma sexual X, en lugar del par habitual (ya sea, dos cromosomas sexuales X o un cromosoma sexual X y un cromosoma sexual Y), se dirá que tiene "monosomía X." La monosomía X también se conoce con el nombre de síndrome de Turner.
¿Qué son las monosomías?
El término monosomía se utiliza para describir la ausencia de uno de los miembros de un par de cromosomas. Por lo tanto, habrá un total de 45 cromosomas en cada célula del cuerpo, en lugar de 46. Por ejemplo, si un bebé nace con un solo cromosoma sexual X, en lugar del par habitual (ya sea, dos cromosomas sexuales X o un cromosoma sexual X y un cromosoma sexual Y), se dirá que tiene "monosomía X." La monosomía X también se conoce con el nombre de síndrome de Turner.
¿Qué es el síndrome de Turner?
El síndrome de Turner es un trastorno genético que se presenta en las niñas y que provoca que sean más bajas que el resto y que no maduren sexualmente a medida que alcanzan la edad adulta. La gravedad de estos problemas varía entre los individuos afectados. También pueden presentarse otros problemas de salud que comprometen al corazón o al aparato renal (es decir, los riñones). Muchos de los problemas que afectan a las niñas con síndrome de Turner pueden controlarse o corregirse con el tratamiento médico adecuado. Este síndrome afecta a una de cada 2.500 niñas recién nacidas.
El nombre "síndrome de Turner" proviene del médico Dr. Henry Turner, quien fue el primero en describir el conjunto de descubrimientos en 1938. No fue sino hasta 1959 que se identificó la causa del síndrome de Turner (la presencia de un sólo cromosoma X).
¿Cuáles son las causas del síndrome de Turner?
Normalmente en la reproducción, el óvulo de la madre y el espermatozoide del padre comienzan teniendo el número habitual de 46 cromosomas. El óvulo y el espermatozoide sufren una división celular en donde los 46 cromosomas se dividen en dos partes iguales y el óvulo y el espermatozoide poseen finalmente 23 cromosomas cada uno. Cuando un espermatozoide con 23 cromosomas fertiliza un óvulo con 23 cromosomas, el bebé tiene finalmente un grupo completo de 46 cromosomas, una mitad obtenida del padre y la otra mitad de la madre.
En ocasiones, ocurre un error durante la formación del óvulo o del espermatozoide, lo que provoca que éste posea un cromosoma sexual menos. Cuando esta célula no puede otorgar el cromosoma sexual al embrión, de manera que existe sólo un cromosoma sexual X, el resultado es el síndrome de Turner. El hecho de tener una sola copia de un cromosoma determinado, en lugar del par habitual, se denomina "monosomía". El síndrome de Turner también se conoce como "monosomía X." El error del cromosoma sexual faltante puede ocurrir tanto en el óvulo como en el espermatozoide; sin embargo, suele ser un error que ocurre en la formación del espermatozoide. No existe nada conocido que el padre pueda haber hecho o no que pudiera haber causado o prevenido la falta de un cromosoma sexual en la formación del espermatozoide. (La probabilidad de que aparezca el síndrome de Turner por lo tanto, no se asocia con la edad de la madre). Las características de este síndrome se originan por la falta de un cromosoma X en cada una de las células del cuerpo.
Alrededor del 50 por ciento de los casos de síndrome de Turner resulta de la monosomía X total. Un tercio tiene dos cromosomas X con parte de una X faltante. Otros tienen un patrón de mosaico (dos o más patrones de cromosoma en las células). Un por ciento pequeño del síndrome de Turner son el resultado de una cantidad normal de cromosomas (46 en total), pero con la falta de una porción del cromosoma X. Cuando falta sólo una parte del cromosoma X (denominado deleción), no todo el cromosoma, las niñas con síndrome de Turner suelen tener características menos pronunciadas del síndrome. Las características del síndrome de Turner presentes dependen de la parte faltante del cromosoma X.
¿Qué clase de problemas tienen generalmente las niñas con síndrome de Turner?
Aproximadamente la mitad de las niñas con síndrome de Turner presentarán al nacer manos y pies hinchados, además de un cuello ancho y alado. Durante la gestación, el médico puede haber detectado una estructura llamada "higroma quístico" al realizar una ecografía fetal. Los higromas quísticos son sacos llenos de líquido ubicados en la base del cuello y suelen desaparecer antes del nacimiento; pero, en ciertos casos, permanecen presentes durante el período neonatal.
Las niñas con síndrome de Turner suelen presentar una línea de nacimiento del cabello baja en la parte posterior del cuello, diferencias mínimas en la forma y posición de las orejas y un tórax ancho con pezones muy separados entre sí, una cantidad mayor de pequeños lunares marrones (nevos) sobre la piel y uñas con un nacimiento profundo. La característica más visible del síndrome de Turner es la baja estatura. La altura promedio de una mujer adulta con síndrome de Turner es de 1,43 m (4 pies, 8 pulgadas). La mayoría de las mujeres con síndrome de Turner nace con los ovarios poco desarrollados o sin ovarios. Los ovarios producen estrógeno y, sin éste, se produce un desarrollo sexual incompleto. Los signos típicos de la pubertad - desarrollo de los senos, menstruación y crecimiento del vello púbico y axilar - no ocurren si no se lleva a cabo un tratamiento hormonal. En la mayoría de los casos, la infertilidad resultante no puede corregirse. En el síndrome de Turner también son comunes los problemas cardíacos, renales y de tiroides, y deben ser evaluados precozmente. Alrededor de una de cada diez niñas con síndrome de Turner nace con coartación de la aorta (constricción de la arteria principal que parte del corazón), la cual suele requerir una corrección quirúrgica. Otras características que se han observado en el síndrome de Turner incluyen problemas de alimentación durante la niñez, infecciones en el oído medio, problemas esqueléticos y "cúbito valgo." El cúbito valgo describe básicamente una situación en la cual la persona parada con los brazos a los lados, presentará los hombros levemente caídos. No es posible mantener los brazos perfectamente derechos a los lados. La diabetes, la piel seca, la presión sanguínea alta, una mandíbula pequeña y un paladar (la parte superior del interior de la boca) angosto y muy arqueado son otros hallazgos médicos del síndrome de Turner.
Las niñas con síndrome de Turner poseen una inteligencia normal. Tienden a obtener puntajes más elevados en su coeficiente intelectual verbal que en el no verbal, y pueden presentar problemas en la percepción espacial y una incidencia mayor de trastornos de aprendizaje específicos.
¿Cómo se diagnostica el síndrome de Turner?
Cuando una niña nace con características que sugieren el síndrome de Turner, se suele tomar una pequeña muestra de sangre y analizar los cromosomas para determinar la ausencia de un cromosoma sexual. A veces, las niñas con síndrome de Turner no muestran problemas en la infancia o en la niñez, y el médico sólo comienza a sospechar que el síndrome de Turner puede estar presente cuando no pueden desarrollarse en la pubertad. Una vez más, no todas las niñas con síndrome de Turner presentan todas las características descriptas aquí. Existe una gran variabilidad. Diagnostican a algunas muchachas con el síndrome de Turner durante el período recién nacido, mientras que otras se diagnostican durante niñez y en su adolescencia.
Las anomalías cromosómicas, como el síndrome de Turner, pueden diagnosticarse frecuentemente antes del nacimiento a través del análisis de las células del líquido amniótico o de la placenta. La ecografía fetal durante la gestación también puede proporcionar información sobre la posibilidad de la presencia del síndrome de Turner, pero la ecografía no tiene una precisión del 100 por ciento, dado que algunos bebés de sexo femenino con el síndrome de Turner presentan en la ecografía el mismo aspecto que un bebé sin problemas. Un análisis cromosómico, ya sea de una muestra de sangre o de células provenientes del líquido amniótico o de la placenta, tiene una exactitud superior al 99,9 por ciento.
¿Cuál es el riesgo de que los padres de una niña con síndrome de Turner tengan otra hija con el síndrome de Turner?
En general, en cada embarazo subsiguiente, la probabilidad de tener otra hija con síndrome de Turner no sería mayor al riesgo de anomalías cromosómicas que tiene que ver con la edad y que afecta a todas las mujeres.
Después del nacimiento, el médico suele extraer una muestra de sangre del bebé que podría padecer el síndrome de Turner para realizar un análisis cromosómico (denominado cariotipo). éste confirma los hallazgos médicos del síndrome de Turner y determina la anomalía cromosómica subyacente. Su médico puede explicarle los resultados del examen o puede derivarlo a un genetista o un asesor genético, quien puede explicar los resultados de los exámenes cromosómicos en detalle y también los exámenes disponibles para diagnosticar los
¿Se puede curar o prevenir el síndrome de Turner?
No existe cura para el síndrome de Turner; sin embargo, muchos de los problemas más serios pueden ser tratados. Por ejemplo, se puede administrar un tratamiento con hormonas de crecimiento y andrógenos para incrementar la talla definitiva del adulto; se puede realizar un tratamiento de reemplazo hormonal para que las niñas desarrollen las características sexuales secundarias; la coartación de la aorta puede corregirse quirúrgicamente si fuese necesario; y existen medicamentos para el tratamiento de la presión sanguínea alta, la diabetes y los problemas de tiroides.
¿Dónde pueden obtener más información las familias afectadas por el síndrome de Turner?
Existen organizaciones en todo el país que brindan información y apoyo a las familias con niñas afectadas por el síndrome de Turner.
Las Anomalías Estructurales:
las Deleciones (Cri du Chat) y las Duplicaciones (Pallister Killian)
¿Cuáles son las anomalías cromosómicas estructurales?
Las anomalías cromosómicas estructurales se presentan cuando hay un cambio en la estructura o en los componentes de un cromosoma. Generalmente el total de cromosomas es normal (46 por célula). Las anomalías cromosómicas estructurales ocurren cuando se pierde parte del cromosoma, cuando hay material cromosómico adicional o cuando dos partes se han intercambiado de lugar. Como consecuencia, esto conduce al exceso o a la carencia de material genético, lo que provoca algunos defectos congénitos.
Cada cromosoma está constituido por varios segmentos que generalmente se clasifican como "brazo corto" y "brazo largo". El brazo corto, la mitad superior del cromosoma, se denomina "brazo p" y el brazo largo, la mitad inferior del cromosoma, es el "brazo q." El centromere es la parte de centro de un cromosoma que aparezca "pellizcado" entre los brazos "p" y "q".
¿Cuáles son las anomalías cromosómicas estructurales?
Las anomalías cromosómicas estructurales se presentan cuando hay un cambio en la estructura o en los componentes de un cromosoma. Generalmente el total de cromosomas es normal (46 por célula). Las anomalías cromosómicas estructurales ocurren cuando se pierde parte del cromosoma, cuando hay material cromosómico adicional o cuando dos partes se han intercambiado de lugar. Como consecuencia, esto conduce al exceso o a la carencia de material genético, lo que provoca algunos defectos congénitos.
Cada cromosoma está constituido por varios segmentos que generalmente se clasifican como "brazo corto" y "brazo largo". El brazo corto, la mitad superior del cromosoma, se denomina "brazo p" y el brazo largo, la mitad inferior del cromosoma, es el "brazo q." El centromere es la parte de centro de un cromosoma que aparezca "pellizcado" entre los brazos "p" y "q".
¿Qué son las deleciones?
El término "deleción" significa simplemente que una parte del cromosoma se perdió o se "eliminó". Un pieza muy pequeña de un cromosoma puede contener muchos genes diferentes. Cuando hay pérdida de material genético, puede haber errores en el desarrollo del bebé, como consecuencia de la pérdida de algunas de las "instrucciones". Un ejemplo de un síndrome genético provocado por deleción es el denominado "Cri du Chat", en el cual ocurre una deleción o pérdida de parte del cromosoma 5.
¿Qué es el Cri du Chat?
Anualmente, el Cri du Chat o "síndrome del maullido de gato" se encuentra en aproximadamente uno en 20.000 a 50.000 nacimientos vivos en los Estados Unidos. La causa del Cri du Chat es la deleción del cromosoma 5p, cuya nomenclatura es "5p-". Las características de los bebés que sufren Cri du Chat son: llanto muy agudo, falta de tonicidad muscular, microcefalia y bajo peso al nacer. También tienen problemas con el lenguaje, y es posible que se expresen a través de una cantidad reducida de palabras o lenguaje de señas. Otros problemas de salud que pueden presentarse incluyen retardo para comenzar a caminar, problemas en la alimentación, hiperactividad, escoliosis y retardo mental severo. El promedio de vida de la mayoría de las personas con Cri du Chat estará dentro de lo normal, a menos que nazcan con otros defectos severos en los órganos.
Para lograr el desarrollo pleno del potencial de un niño con Cri du Chat, es importante la educación desde una edad temprana, además de terapias físicas y del lenguaje.
¿Qué son las duplicaciones?
El término "duplicación" significa simplemente que una parte del cromosoma está duplicada o presenta dos copias. El resultado es el material genético adicional, aun cuando el total de cromosomas está generalmente dentro de lo normal. Dado que una pieza muy pequeña de un cromosoma puede contener muchos genes diferentes, el material genético presente en una duplicación puede provocar que dichos genes no funcionen correctamente. Como consecuencia de estas "instrucciones adicionales", pueden producirse errores en el desarrollo de un bebé. Una manera de pensar en la duplicación es pensar que los 46 cromosomas forman un libro de cocina, y cada uno de los cromosomas es una receta. Si una deleción es un ingrediente que falta en una receta, un duplicación es un ingrediente adicional. Un ejemplo de un síndrome genético provocado por duplicación es el denominado "síndrome de Pallister Killian", en el cual parte del cromosoma 12 está duplicado.
¿Qué es el síndrome de Pallister Killian?
El síndrome de Pallister Killian es el resultado del material genético adicional del cromosoma 12. En general, se presenta una mezcla de células (mosaicismo), algunas con material adicional del cromosoma 12 y otras normales (46 cromosomas sin material genético adicional del cromosoma 12). Los bebés que sufren este síndrome padecen muchos problemas, entre los que se incluyen retardo mental severo, falta de tonicidad muscular, facciones toscas y frente prominente. Entre otras características se pueden mencionar el labio superior muy delgado, el labio inferior más grueso y la nariz corta. Entre otros problemas de salud que ocasiona este síndrome se incluyen convulsiones, mala alimentación, rigidez en las articulaciones, cataratas (en los adultos), pérdida auditiva y defectos cardíacos. El promedio de vida de las persona que padecen síndrome de Pallister Killian es reducido, aunque pueden vivir más de 40 años.
Las Translocaciones
¿Qué son las translocaciones?
El término translocación se utiliza cuando se presentan modificaciones en la ubicación de determinado material cromosómico. Existen dos tipos de translocaciones: recíproca y robertsoniana. En una translocación recíproca, dos cromosomas diferentes intercambian segmentos entre sí.
En una translocación robertsoniana, un cromosoma completo se adhiere a otro en el centrómero. El centromere es la parte de centro de un cromosoma que aparezca "pellizcado" entre los brazos "p" y "q".
Este nuevo cromosoma que se forma se denomina cromosoma por translocación. La translocación de este ejemplo se encuentra entre los cromosomas 14 y 21. Cuando un bebé nace con este tipo de cromosoma por translocación (entre el 14 y el 21), además de un cromosoma 14 normal y dos cromosomas 21 normales, el bebé sufrirá síndrome de Down, también denominado síndrome de Down por translocación.
El Síndrome de Down por Translocación
¿Qué es el síndrome de Down por translocación?
El síndrome de Down por translocación hace referencia al reordenamiento del material cromosómico. Existen tres cromosomas 21, al igual que en la trisomía 21, pero uno de ellos está adherido a otro cromosoma, en lugar de estar separado. El cromosoma 21 adicional es el que provoca los problemas que constituyen el síndrome de Down. En el síndrome de Down por translocación, el cromosoma 21 adicional puede adherirse al cromosoma 14, o al 13, 15 o 22. En algunos casos, dos cromosomas 21 pueden adherirse uno a otro.
Entre el 3 y el 4 por ciento de los bebés que nacen con síndrome de Down tienen síndrome de Down por translocación. En una habitación con 100 bebés con síndrome de Down, todos los bebés se parecerán entre sí y tendrán características y problemas de salud similares. No será fácil detectar los 3 o 4 que presenten la translocación.
Cada vez que se encuentra una translocación en un niño, se estudian los cromosomas de los padres para determinar si dicha translocación es hereditaria o no. Si uno de los padres presenta un cromosoma por translocación, entonces el médico concluye que el bebé heredó ese cromosoma de su padre. En realidad, el padre tendrá un total de 45 cromosomas en cada célula de su cuerpo, cuyo estado será normal y saludable debido a que aún tienen sólo dos copias de cada cromosoma. Cuando una persona presenta un reordenamiento del material cromosómico, sin material cromosómico extra o ausente, se dice que tiene una "translocación balanceada" o que es un "portador de translocación balanceada".
Los padres con translocaciones balanceadas pueden presentar problemas de fertilidad (problemas para gestar), abortos espontáneos o mayor probabilidad de tener un hijo con problemas de salud. Aunque el padre puede donar la cantidad adecuada de material genético (23 cromosomas) para lograr el embarazo, corre el riesgo de que este material donado sea demasiado o insuficiente. Esto no puede controlarse ni predecirse. Las probabilidades dependen del tipo de reordenamiento cromosómico y de los cromosomas involucrados. Por ejemplo, si la translocación está entre los cromosomas 14 y 21, las probabilidades de síndrome de Down en el embarazo son de entre un 10 y un 15 por ciento si la madre es la portadora de la translocación, y de entre un 3 y un 5 por ciento si el padre es el portador de la translocación. Las probabilidades son diferentes para hombres y mujeres debido a que los espermatozoides y los óvulos se producen de manera diferente. La mujer nace con todos los óvulos que tendrá a lo largo de su vida, mientras que el hombre produce espermatozoides nuevos constantemente.
Existe otro factor importante a tener en cuenta cuando se encuentra una translocación en uno de los padres. Los familiares de los padres (hermanos, hermanas) también pueden haber heredado la translocación y, por lo tanto, pueden correr los mismos riesgos en un embarazo. Por estos motivos, se recomienda que las personas con reordenamientos cromosómicos compartan esta información con sus familiares para que estos puedan realizarse los estudios correspondientes.
¿Qué es el síndrome de Down por translocación?
El síndrome de Down por translocación hace referencia al reordenamiento del material cromosómico. Existen tres cromosomas 21, al igual que en la trisomía 21, pero uno de ellos está adherido a otro cromosoma, en lugar de estar separado. El cromosoma 21 adicional es el que provoca los problemas que constituyen el síndrome de Down. En el síndrome de Down por translocación, el cromosoma 21 adicional puede adherirse al cromosoma 14, o al 13, 15 o 22. En algunos casos, dos cromosomas 21 pueden adherirse uno a otro.
Entre el 3 y el 4 por ciento de los bebés que nacen con síndrome de Down tienen síndrome de Down por translocación. En una habitación con 100 bebés con síndrome de Down, todos los bebés se parecerán entre sí y tendrán características y problemas de salud similares. No será fácil detectar los 3 o 4 que presenten la translocación.
Cada vez que se encuentra una translocación en un niño, se estudian los cromosomas de los padres para determinar si dicha translocación es hereditaria o no. Si uno de los padres presenta un cromosoma por translocación, entonces el médico concluye que el bebé heredó ese cromosoma de su padre. En realidad, el padre tendrá un total de 45 cromosomas en cada célula de su cuerpo, cuyo estado será normal y saludable debido a que aún tienen sólo dos copias de cada cromosoma. Cuando una persona presenta un reordenamiento del material cromosómico, sin material cromosómico extra o ausente, se dice que tiene una "translocación balanceada" o que es un "portador de translocación balanceada".
Los padres con translocaciones balanceadas pueden presentar problemas de fertilidad (problemas para gestar), abortos espontáneos o mayor probabilidad de tener un hijo con problemas de salud. Aunque el padre puede donar la cantidad adecuada de material genético (23 cromosomas) para lograr el embarazo, corre el riesgo de que este material donado sea demasiado o insuficiente. Esto no puede controlarse ni predecirse. Las probabilidades dependen del tipo de reordenamiento cromosómico y de los cromosomas involucrados. Por ejemplo, si la translocación está entre los cromosomas 14 y 21, las probabilidades de síndrome de Down en el embarazo son de entre un 10 y un 15 por ciento si la madre es la portadora de la translocación, y de entre un 3 y un 5 por ciento si el padre es el portador de la translocación. Las probabilidades son diferentes para hombres y mujeres debido a que los espermatozoides y los óvulos se producen de manera diferente. La mujer nace con todos los óvulos que tendrá a lo largo de su vida, mientras que el hombre produce espermatozoides nuevos constantemente.
Existe otro factor importante a tener en cuenta cuando se encuentra una translocación en uno de los padres. Los familiares de los padres (hermanos, hermanas) también pueden haber heredado la translocación y, por lo tanto, pueden correr los mismos riesgos en un embarazo. Por estos motivos, se recomienda que las personas con reordenamientos cromosómicos compartan esta información con sus familiares para que estos puedan realizarse los estudios correspondientes.
Otros Arreglos Cromosómicos: Anillos e Inversiones
¿Qué es una inversión?
Existen diversas formas en las que puede alterarse la estructura de un cromosoma. Una de ellas se denomina "inversión". El término "inversión" se utiliza cuando un cromosoma se fragmenta en dos puntos y el segmento intermedio gira al revés y luego vuelve a unirse.
Algunas inversiones son tan frecuentes en la población que no es necesario realizar una prueba adicional para detectarlas. Si se encuentra una inversión rara o poco frecuente en un niño, se estudian los cromosomas de los padres para determinar si dicha inversión es hereditaria o no. Si uno de los padres presenta esta inversión, entonces el médico concluye que el bebé heredó dicha inversión de su padre. El hecho de que generalmente el estado del padre sea normal y saludable prueba que la inversión no provocó ningún daño genético en el momento en que el cromosoma se fragmentó y giró al revés. (Los demás familiares de los padres también pueden haber heredado la inversión y pueden decidir si realizar o no un estudio cromosómico.) Sin embargo, si la inversión no se presenta en alguno de los padres, existe la posibilidad de que parte del material genético se haya perdido o alterado en el reordenamiento. Esto podría provocar problemas en el desarrollo del bebé, incluyendo defectos congénitos.
¿Qué es un cromosoma anular?
Otro ejemplo de reordenamiento cromosómico es lo que se denomina cromosoma "anular". El término cromosoma "anular" se utiliza para describir un cromosoma cuyas extremidades se fragmentaron y se unieron para formar un círculo o "anillo".
Debido a la rotura de las extremidades, la información genética se pierde. Los anillos también pueden formarse sin que las extremidades se rompan: el brazo p y el brazo q se "pegan" sin pérdida aparente de material genético. En algunos casos, los anillos (independientemente de cómo se originaron) provocan problemas en el desarrollo del bebé. Esto se debe a que el anillo interfiere en la división celular y puede provocar que los genes no funcionen de manera adecuada debido a su reordenamiento.
Los cromosomas anulares no siempre ocasionan problemas de salud o retrasos en el desarrollo; todo depende del tamaño del anillo, cuánto material genético se perdió y cuántos cromosomas están involucrados. Si se encuentra un cromosoma anular en un niño, se estudian los cromosomas de los padres para determinar si dicho anillo es hereditario o no. Si uno de los padres presenta un cromosoma anular, entonces el médico concluye que el bebé heredó dicho cromosoma de su padre. Si el estado del padre es normal y saludable, se considera que el anillo no provocó ningún daño genético en el momento en que se formó el cromosoma. (Los demás familiares de los padres también pueden haber heredado el cromosoma anular y pueden decidir si realizar o no un estudio cromosómico.) Sin embargo, si el anillo no se presenta en alguno de los padres, existe la posibilidad de que parte del material genético se haya perdido o alterado en el reordenamiento. Esto podría provocar problemas en el desarrollo del bebé, incluyendo defectos congénitos.
¿Qué es una inversión?
Existen diversas formas en las que puede alterarse la estructura de un cromosoma. Una de ellas se denomina "inversión". El término "inversión" se utiliza cuando un cromosoma se fragmenta en dos puntos y el segmento intermedio gira al revés y luego vuelve a unirse.
Algunas inversiones son tan frecuentes en la población que no es necesario realizar una prueba adicional para detectarlas. Si se encuentra una inversión rara o poco frecuente en un niño, se estudian los cromosomas de los padres para determinar si dicha inversión es hereditaria o no. Si uno de los padres presenta esta inversión, entonces el médico concluye que el bebé heredó dicha inversión de su padre. El hecho de que generalmente el estado del padre sea normal y saludable prueba que la inversión no provocó ningún daño genético en el momento en que el cromosoma se fragmentó y giró al revés. (Los demás familiares de los padres también pueden haber heredado la inversión y pueden decidir si realizar o no un estudio cromosómico.) Sin embargo, si la inversión no se presenta en alguno de los padres, existe la posibilidad de que parte del material genético se haya perdido o alterado en el reordenamiento. Esto podría provocar problemas en el desarrollo del bebé, incluyendo defectos congénitos.
¿Qué es un cromosoma anular?
Otro ejemplo de reordenamiento cromosómico es lo que se denomina cromosoma "anular". El término cromosoma "anular" se utiliza para describir un cromosoma cuyas extremidades se fragmentaron y se unieron para formar un círculo o "anillo".
Debido a la rotura de las extremidades, la información genética se pierde. Los anillos también pueden formarse sin que las extremidades se rompan: el brazo p y el brazo q se "pegan" sin pérdida aparente de material genético. En algunos casos, los anillos (independientemente de cómo se originaron) provocan problemas en el desarrollo del bebé. Esto se debe a que el anillo interfiere en la división celular y puede provocar que los genes no funcionen de manera adecuada debido a su reordenamiento.
Los cromosomas anulares no siempre ocasionan problemas de salud o retrasos en el desarrollo; todo depende del tamaño del anillo, cuánto material genético se perdió y cuántos cromosomas están involucrados. Si se encuentra un cromosoma anular en un niño, se estudian los cromosomas de los padres para determinar si dicho anillo es hereditario o no. Si uno de los padres presenta un cromosoma anular, entonces el médico concluye que el bebé heredó dicho cromosoma de su padre. Si el estado del padre es normal y saludable, se considera que el anillo no provocó ningún daño genético en el momento en que se formó el cromosoma. (Los demás familiares de los padres también pueden haber heredado el cromosoma anular y pueden decidir si realizar o no un estudio cromosómico.) Sin embargo, si el anillo no se presenta en alguno de los padres, existe la posibilidad de que parte del material genético se haya perdido o alterado en el reordenamiento. Esto podría provocar problemas en el desarrollo del bebé, incluyendo defectos congénitos.
El Mosaicismo
¿Qué es el mosaicismo?
El término mosaicismo se utiliza para describir la presencia de más de un tipo de célula en un individuo. Por ejemplo, una persona puede tener algunas de las células de su cuerpo con 46 cromosomas, mientras que otras células de su cuerpo pueden tener 47 cromosomas. Un ejemplo de mosaicismo en el síndrome de Down con alteración cromosómica en mosaico.
Aproximadamente el 95 por ciento de las personas que padecen síndrome de Down tienen trisomía 21, es decir, un cromosoma 21 adicional en cada célula de su cuerpo. Entre un 3 y un 4 por ciento de los individuos con síndrome de Down padecen síndrome de Down por translocación. Esto significa que el cromosoma 21 adicional, o parte de él, está adosado a otro cromosoma. El 1 y 2 por ciento restante de individuos que padecen síndrome de Down tienen una alteración cromosómica en mosaico. Por consiguiente, se deduce que tienen por lo menos dos tipos de células; algunas con el número normal de cromosomas (un total de 46) y otras con un cromosoma 21 adicional (un total de 47). En raras oportunidades, una persona puede tener más de dos tipos de líneas celulares.
El mosaicismo generalmente se describe por medio de un porcentaje. Por ejemplo, cuando nace un bebé con síndrome de Down, el médico le toma una muestra de sangre para realizar un estudio cromosómico. Por lo general, se analizan 20 células diferentes. Si cinco de esas 20 células son normales (46 cromosomas) y las 15 restantes tienen un cromosoma 21 adicional (47 cromosomas), se determina que el bebé tiene síndrome de Down con alteración cromosómica en mosaico. Dado que el porcentaje de células con un cromosoma adicional es 15 sobre un total de 20, se establecerá que el bebé tiene un nivel de mosaicismo del 75 por ciento. Los porcentajes pueden variar en las distintas partes del cuerpo. El porcentaje de células trisómicas en el músculo puede ser distinto del porcentaje registrado en el cerebro o el porcentaje registrado en la sangre o la piel.
Generalmente, un bebé recibe la mitad de sus cromosomas de cada progenitor, de manera que cada óvulo y espermatozoide tiene 23 cromosomas. Luego de la fertilización, ocurre un proceso rápido de división celular por el cual una célula duplica sus cromosomas (realiza una copia de ellos) y luego se divide por la mitad. En otras palabras, la célula pasa de tener 46 cromosomas a tener 92 cromosomas y luego se divide por la mitad, resultando en dos células nuevas de 46 cromosomas cada una. Este tipo de división celular se denomina mitosis.
En algún momento posterior a la fertilización, se puede producir un error en la mitosis, es decir, una célula tal vez no se divida exactamente en dos mitades. Como consecuencia de ello, algunas células tendrán el número normal o 46 cromosomas mientras que otras tendrán un cromosoma 21 adicional o 47 cromosomas.
¿Qué es el mosaicismo?
El término mosaicismo se utiliza para describir la presencia de más de un tipo de célula en un individuo. Por ejemplo, una persona puede tener algunas de las células de su cuerpo con 46 cromosomas, mientras que otras células de su cuerpo pueden tener 47 cromosomas. Un ejemplo de mosaicismo en el síndrome de Down con alteración cromosómica en mosaico.
Aproximadamente el 95 por ciento de las personas que padecen síndrome de Down tienen trisomía 21, es decir, un cromosoma 21 adicional en cada célula de su cuerpo. Entre un 3 y un 4 por ciento de los individuos con síndrome de Down padecen síndrome de Down por translocación. Esto significa que el cromosoma 21 adicional, o parte de él, está adosado a otro cromosoma. El 1 y 2 por ciento restante de individuos que padecen síndrome de Down tienen una alteración cromosómica en mosaico. Por consiguiente, se deduce que tienen por lo menos dos tipos de células; algunas con el número normal de cromosomas (un total de 46) y otras con un cromosoma 21 adicional (un total de 47). En raras oportunidades, una persona puede tener más de dos tipos de líneas celulares.
El mosaicismo generalmente se describe por medio de un porcentaje. Por ejemplo, cuando nace un bebé con síndrome de Down, el médico le toma una muestra de sangre para realizar un estudio cromosómico. Por lo general, se analizan 20 células diferentes. Si cinco de esas 20 células son normales (46 cromosomas) y las 15 restantes tienen un cromosoma 21 adicional (47 cromosomas), se determina que el bebé tiene síndrome de Down con alteración cromosómica en mosaico. Dado que el porcentaje de células con un cromosoma adicional es 15 sobre un total de 20, se establecerá que el bebé tiene un nivel de mosaicismo del 75 por ciento. Los porcentajes pueden variar en las distintas partes del cuerpo. El porcentaje de células trisómicas en el músculo puede ser distinto del porcentaje registrado en el cerebro o el porcentaje registrado en la sangre o la piel.
Generalmente, un bebé recibe la mitad de sus cromosomas de cada progenitor, de manera que cada óvulo y espermatozoide tiene 23 cromosomas. Luego de la fertilización, ocurre un proceso rápido de división celular por el cual una célula duplica sus cromosomas (realiza una copia de ellos) y luego se divide por la mitad. En otras palabras, la célula pasa de tener 46 cromosomas a tener 92 cromosomas y luego se divide por la mitad, resultando en dos células nuevas de 46 cromosomas cada una. Este tipo de división celular se denomina mitosis.
En algún momento posterior a la fertilización, se puede producir un error en la mitosis, es decir, una célula tal vez no se divida exactamente en dos mitades. Como consecuencia de ello, algunas células tendrán el número normal o 46 cromosomas mientras que otras tendrán un cromosoma 21 adicional o 47 cromosomas.
Síndrome de Down con Alteración Cromosómica en Mosaico
¿Qué es el síndrome de Down con alteración cromosómica en mosaico?
¿Qué es el síndrome de Down con alteración cromosómica en mosaico?
El mosaicismo, término que se utiliza para describir la presencia de más de un tipo de célula en un individuo, generalmente se describe por medio de un porcentaje. Por ejemplo, cuando nace un bebé con síndrome de Down, el médico le toma una muestra de sangre para realizar un estudio cromosómico. Por lo general, se analizan 20 células diferentes. Si cinco de esas 20 células son normales (46 cromosomas) y las 15 restantes tienen un cromosoma 21 adicional (47 cromosomas), se determina que el bebé tiene síndrome de Down con alteración cromosómica en mosaico. Dado que el porcentaje de células con un cromosoma adicional es 15 sobre un total de 20, se dirá que el bebé tiene un nivel de mosaicismo del 75 por ciento. Los porcentajes pueden variar en las distintas partes del cuerpo. El porcentaje de células trisómicas en el músculo puede ser distinto del porcentaje registrado en el cerebro o el porcentaje registrado en la sangre o la piel.
Clínicamente, los bebés que nacen con síndrome de Down por alteración cromosómica en mosaico presentan las mismas características y problemas de salud que los bebés que nacen con trisomía 21 o con síndrome de Down por translocación. Sin embargo, la presencia de células con un número normal de cromosomas (46) puede contribuir a la disminución o bien a la presentación menos severa de las características propias del síndrome de Down.
¿Existen estudios que permitan comprender mejor el síndrome de Down con alteración cromosómica en mosaico?
Se están llevando a cabo estudios que intentan determinar si existe alguna diferencia en la salud de individuos con síndrome de Down por alteración cromosómica en mosaico en función de la manera en la que se produjo el mosaicismo. Además, se han realizado estudios para determinar si el porcentaje de mosaicismo puede predecir el resultado clínico, tal como el coeficiente intelectual (su sigla en inglés es IQ) del individuo o las probabilidades de que sufra algún defecto cardíaco. Estos estudios demostraron que el porcentaje de mosaicismo no sirve para pronosticar un resultado con exactitud. Las características de las personas con síndrome de Down con alteración cromosómica en mosaico y, por ende, con trisomía de las células 21 pueden variar enormemente, ya que algunas presentan características muy leves y otras manifiestan la mayoría de las características propias del síndrome de Down.
¿Cuáles son las posibilidades de que se produzca un nuevo caso de síndrome de Down con alteración cromosómica en mosaico en la familia?
En general, el riesgo de recurrencia del síndrome de Down con alteración cromosómica en mosaico es menor al 1 por ciento, hasta la edad materna de 35, en el que aumenta considerablemente. El riesgo de que se produzcan anomalías cromosómicas durante un embarazo se determina en función de la edad de la madre al momento de dar a luz y aumentos de la edad de la madre anticipos .
Cómo Ocurren las Anomalías Cromosómicas: la Meiosis, la Mitosis, la Edad Materna, el Entorno
¿Cómo ocurren las anomalías cromosómicas?
Las anomalías cromosómicas generalmente ocurren como consecuencia de uno, o más, de los siguientes factores:
MeiosisPor lo general, las anomalías cromosómicas ocurren como consecuencia de un error producido en la división celular. Meiosis es el término que se utiliza para describir la división celular por la que atraviesan el óvulo y el espermatozoide durante el desarrollo. Habitualmente, la meiosis causa la división del material cromosómico, de manera que cada padre aporte 23 cromosomas a un embarazo:
Esto resulta en un óvulo o un espermatozoide que sólo tiene 23 cromosomas. Cuando se produce la fertilización, se origina el número total normal de 46 cromosomas. Si la meiosis no se produce adecuadamente, un óvulo o un espermatozoide podría terminar con demasiados cromosomas o con una cantidad insuficiente de estos últimos. Luego de la fertilización, el bebé puede recibir un cromosoma adicional (llamado trisomía) o tener un cromosoma en falta (llamado monosomía):
Si bien los embarazos que presentan una trisomía o una monosomía pueden llegar a término y dar a luz a un niño con problemas de salud, también es posible que se produzca un aborto espontáneo o que el bebé nazca muerto, debido a la anomalía cromosómica. En estudios realizados en abortos espontáneos durante el primer trimestre, en aproximadamente un 60 por ciento (o más) de los casos se trataba de alguna anomalía cromosómica. En estudios realizados en bebés nacidos muertos, entre un 5 y un 10 por ciento de los casos presentaba alguna anomalía cromosómica.
MitosisMitosis es el término que se utiliza para describir la división celular por la que atraviesan todas las demás células, además del óvulo y el espermatozoide, durante el desarrollo. Por lo general, la mitosis causa una duplicación, seguida de una división del material cromosómico, de manera que cada una de las células duplica el número de cromosomas hasta llegar a 92 y luego se divide por la mitad, resultando en la cantidad total normal de 46 cromosomas. La mitosis comienza luego de la fertilización:
Este proceso se repite hasta que el bebé se desarrolla por completo. La mitosis continúa de por vida para regenerar las células de la piel, los glóbulos y otros tipos de células que se dañan o, simplemente, se mueren.
Durante el embarazo, se puede producir un error en la mitosis tal como se describió anteriormente en el caso de la meiosis. Si los cromosomas no se dividen en mitades exactas, las células nuevas pueden tener un cromosoma adicional (un total de 47) o un cromosoma faltante (un total de 45). Ésta es otra manera por la que un bebé puede nacer con una anomalía cromosómica. Los errores de mitosis son responsables de algunos casos de mosaicismo.
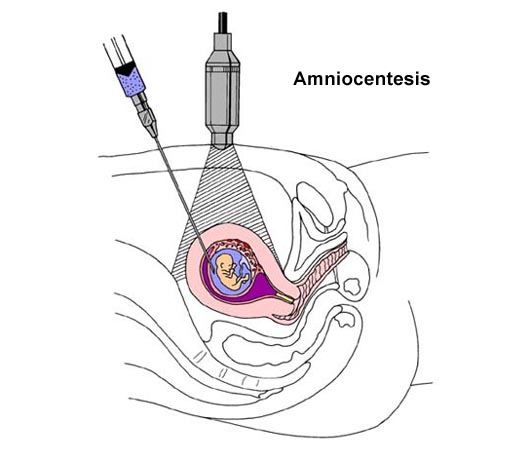
Edad maternaSi una madre planea dar a luz a los 35 años de edad (o más), es posible que se le aconseje realizar algún tipo de asesoramiento genético o diagnóstico prenatal, como por ejemplo una amniocentesis, debido a su edad. Existe una diferencia en la forma en que se fabrican los óvulos y los espermatozoides. Las mujeres nacen con todos los óvulos que permanecerán con ellas a lo largo de toda la vida. Con el paso del tiempo, la cantidad de óvulos en sus ovarios disminuye paulatinamente. Por ende, si una mujer tiene 35 años de edad, los óvulos disponibles en sus ovarios también tendrán la misma edad. El riesgo de tener un bebé con alguna anomalía cromosómica aumenta con la edad de la madre. Algunos científicos y médicos creen que esto se debe al envejecimiento de los óvulos y a un número incorrecto de cromosomas al momento de la fertilización. Como consecuencia del proceso de envejecimiento, existen mayores probabilidades de que se produzcan errores en la meiosis.
Por otro lado, los hombres producen espermatozoides continuamente. Por consiguiente, un hombre puede tener 35 años de edad, pero su esperma no coincide con la edad mencionada. Por ende, no existe un aumento en el riesgo de que se produzcan anomalías cromosómicas en función de la edad del padre. Esto no significa que los errores cromosómicos no se produzcan en el espermatozoide. Se producen, sólo que los errores no se vinculan con la edad del padre.
EntornoMuchos padres que han tenido un niño con alguna anomalía cromosómica investigan durante toda sus vidas y se preguntan si existe algún tipo de relación entre las distintas exposiciones ambientales que han sufrido año tras año y el hecho de haber dado a luz a un bebé con una anomalía cromosómica. Hasta la fecha, no se ha determinado la presencia de ningún agente específico en el entorno, ya sea los rayos X, los medicamentos, los alimentos, los hornos microondas, etc., que pudiese contribuir con el nacimiento de un bebé con una anomalía cromosómica. En realidad, la mayoría de los padres que tienen un niño con una anomalía cromosómica no presenta hábitos, estilos de vida o exposiciones diferentes a los de los padres cuyos hijos no padecen una anomalía cromosómica.
Cada vez existe más evidencia que indica que la manera en la que el organismo de una mujer procesa el ácido fólico de la vitamina B podría estar relacionada con la ocurrencia de anomalías cromosómicas. Las mujeres que no procesan esta vitamina en forma completa pueden tener cierta predisposición a tener un niño con alguna anomalía cromosómica. Esto aún no ha sido probado, pero sabiendo que se trata de una posibilidad, las mujeres en edad reproductiva tienen un buen motivo para tomar una multivitamina que contenga ácido fólico antes de quedar embarazadas y vitaminas prenatales durante el embarazo para reducir este riesgo potencial.

Casino Review for 2021 – 100% up to €1000
ResponderEliminarCasino. 광주광역 출장샵 Casino Review: Bonuses, Payouts, Features. Experience 서울특별 출장마사지 the 안산 출장마사지 benefits of being part of 구미 출장마사지 a trusted brand. Get the most out of your 양주 출장마사지